
SOP-Guard
Der Effizienz-Booster
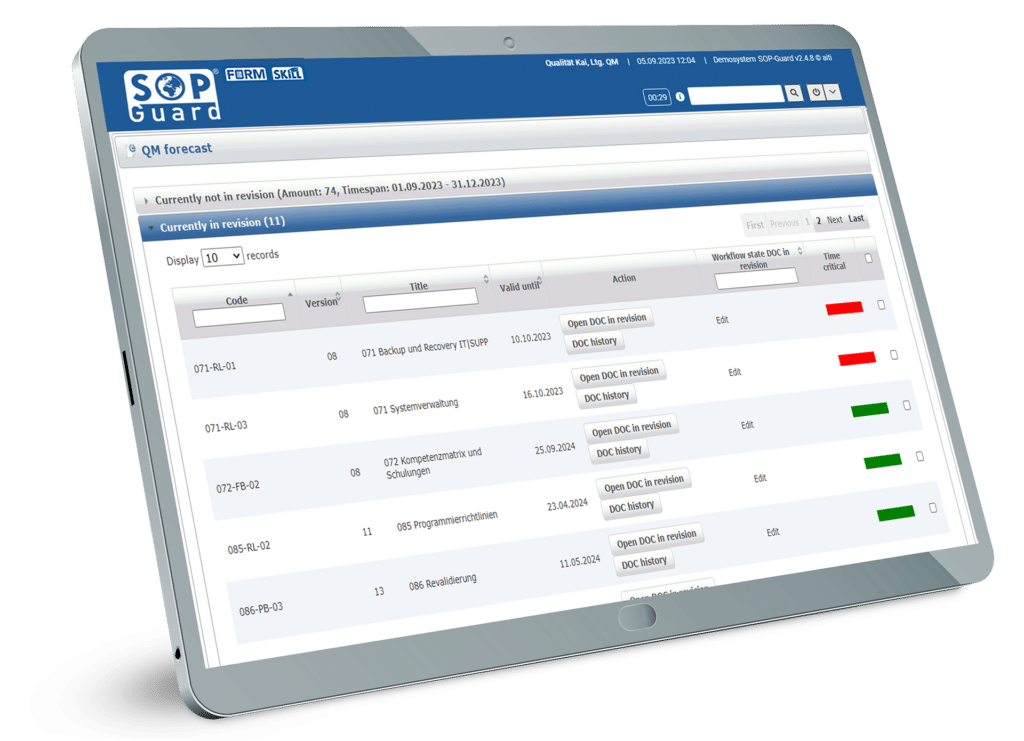
SOP-Guard Dokumentverwaltung
Entfalten Sie das volle Potenzial Ihrer administrativen Aufgaben im Qualitätsmanagement und optimieren Sie Ihre Workflows mit der innovativen Unterstützung von SOP-Guard. Unabhängig von Ihrer Branche ermöglicht es SOP-Guard Ihnen, Dokumente mühelos zu erstellen, zu überprüfen, freizugeben, zu verteilen und zu archivieren. Unsere erfolgsorientierte Dokumentenschulungsfunktion ermöglicht Ihnen eine nutzerbasierte Kontrolle über Ihr elektronisches Dokumentenmanagement für GxP-Systeme. Durch die Nutzung von SOP-Guard ebnet sich der Weg für eine Zukunft der Effizienz, Produktivität und des Erfolgs.



Top 5 Vorteile
- Validierte Software für eine 100 % elektronische Verwaltung von Dokumenten in regulierten Unternehmen
- Exaktes Monitoring von Fristen inklusive zeitgerechter Korrekturmaßnahmen.
- Korrekte Abbildung von Zuständigkeiten und Verantwortlichkeiten.
- Automatisierte Zuordnung von Dokumenten an die richtige Usergruppe.
- Automatisierung bislang manueller Arbeitsschritte in der Dokumentenverwaltung.
SOP-Guard ist validiert nach GAMP 5,
GxP- und FDA konform.
Kundenstimmen
Der geringe Aufwand bei Erstellung, Prüfung, Genehmigung, Verwaltung und Schulung der unterschiedlichsten Vorgabedokumente sowie die Flexibilität von SOP-Guard waren klare Pluspunkte.
Dr. Martina MaritzHead Corporate Quality Assurance Develco Pharma GmbH
Ein DMS kann drei Viertel unserer Anforderungen abdecken, aber die Würze liegt in den restlichen 25 Prozent, die uns SOP-Guard bietet.
Ivica SaricLeiter IT | Validierungskoordinator ITUrsapharm Arzneimittel GmbH
SOP-Guard ist einfach zu bedienen und schnell zu verstehen. Das hat den Roll-Out in sechs osteuropäische Länder erheblich vereinfacht.
Mag.pharm. Georg E. SchmidtHead of Pharmaceutical AffairsChiesi Pharmaceuticals GmbH
SOP Guard erleichtert uns das Leben in einer kleineren akademischen GMP-Einrichtung enorm. Einfache Bedienung, logischer Aufbau, rascher Kundenservice und gut vorbereitete Unterlagen für die Validierung und Revalidierung des Systems machen es zu einem sehr hilfreichen Programm für die effiziente und GMP-gerechte Dokumentenlenkung.
Dr. Daniela WehlerSachkundige PersonIII. Med. Klinik und Poliklinik, Universitätsmedizin Mainz
Quick-Facts
SOP-Guard wurde speziell für GxP-Umgebungen entwickelt und bietet eine zentrale Plattform für einfaches Management und Kontrolle wichtiger Dokumente. Die spezialisierten und leistungsstarken Programmfunktionen in Verbindung mit einer intuitiven Benutzeroberfläche ermöglichen eine Reduzierung von Fehlern und Zeitverlust. SOP-Guard ist vollständig konform mit aktuellen regulatorischen Anforderungen und bietet somit verbesserte Effizienz, erhöhte Sicherheit und bessere Dokumentenkontrolle.
SOP-Guard optimiert manuelle, zeitaufwändige Dokumentenprozesse und sorgt für eine reibungslose Strukturierung. Dabei ermöglicht unser System eine effiziente Dokumentenverwaltung mit schneller Datenabfrage, wodurch wertvolle Zeitressourcen eingespart werden können. Wir garantieren eine schnelle und produktive Implementierung, kurze Schulungszeiten und eine flache Einarbeitungskurve.
Wir legen großen Wert auf das Feedback unserer Benutzer und Kunden, das aktiv in die Entwicklung unserer Software integriert wird. Unsere Lösung hat zahlreiche positive Bewertungen erhalten und erfolgreich Audits und Inspektionen bestanden. Lassen Sie uns Ihnen helfen, Ihre Dokumentenmanagementprozesse für den Erfolg Ihres Unternehmens zu verbessern.
Unsere Softwarelösung bietet die Installation an einem Ort Ihrer Wahl (vor Ort, private Cloud …) die sicherstellt, dass sowohl die Software als auch die Datenspeicherung innerhalb des Netzwerks Ihres Unternehmens stattfinden. Entwickelt mit Web-Technologie ist unsere Software kompatibel mit gängigen Geräten wie PCs, Laptops und Tablets. Die Software wird Ihnen als validierte virtuelle Maschine geliefert und unser Wartungsvertrag legt streng unsere Verantwortlichkeiten fest und klärt jeweilige Rollen. Zusätzlich bieten wir zahlreiche Schnittstellen, die eine nahtlose Konnektivität mit bestehenden Systemen ermöglichen (z.B. Active Directory via LDAP).
– SOP-Guard ist als Produkt gemäß GAMP 5 (Kategorie 3) vollumfänglich validiert.
– Die Software entspricht den Anforderungen u. a. folgender Regularien: GxP (EU), ICH sowie 21 CFR Part 11.
– Nachverfolgbarkeit durch systeminternen Audit-Trail zur detaillierten Aufzeichnung von Aktionen im System
– Umfangreiche Validierungspakete (Installationsdokumentation, Testfalldokumente, Risikoanalyse …)
SOP-Guard schnell erklärt
Downloads
SOP-Guard
| Titel | Kategorien | Aktualisierungsdatum | Download |
|---|---|---|---|
|
SOP Guard: Dokumentenmanagement – modern & GxP konform |
Softwarelösungen, SOP-Guard | 19. September 2023 | Download |


